
Genome analysis from the Mason Lab, Weill Cornell Medicine (WCM)
(See also these slides for an overall report)
I. Ancestry
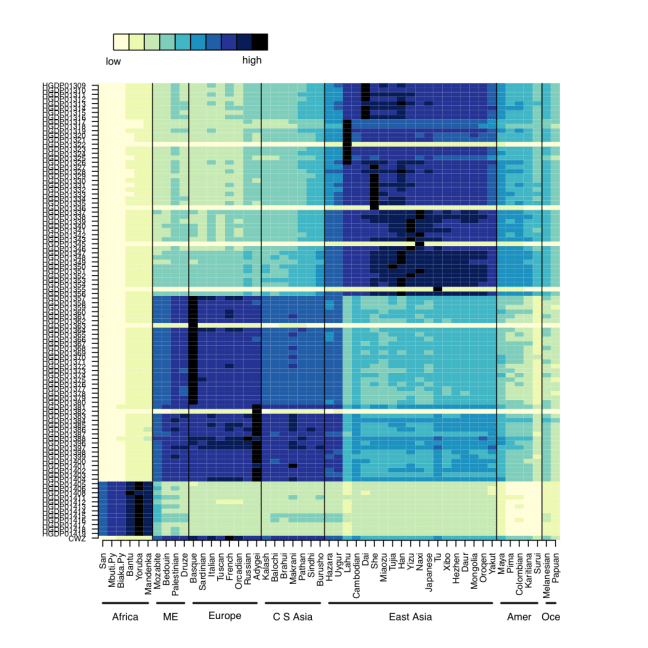
We first confirmed that CZ was indeed genetically male, based on the X-chromosome’s Loss of Heterozygosity (LOH), and this indeed was true (>98% homozygous SNV calls). Next, we wanted to look at the ancestry of CZ using Ancestry Mapper, a tool that allows you to calculate the genetic distance of a person’s genome relative to 51 reference populations, using over 1,000 individuals from the Human Genome Diversity Project. Interestingly, we also used this same algorithm to map the DNA left on subway surfaces with the neighborhood’s U.S. Census data (Black, White, Asian, Hispanic) and showed that neighborhoods’ DNA that often matched the Census data (Afshinnekoo et al., 2015). But, for CZ (bottom row, Figure 1), we found him closest to the French, Italian, and Basque reference populations, but also with similarity to European and Middle Eastern ancestry.
Figure 1- Predicted ancestry of CZ genome. Genetic similarities are plotted for a large set of reference populations (x-axis) from different regions of the world, including Africa, Middle East, Europe, Central South Asia, East Asia, Americas, and Oceania. CZ’s genome is at the bottom.
References :
Afshinnekoo E, Meydan C, Chowdhury S, Jaroudi D, et al. Mason CE. “Geospatial Resolution of Human and Bacterial Diversity from City-scale Metagenomics.” Cell Systems. 2015 Jul 29;1(1):72-87. PMC4651444.
Magalhães TR, Casey JP, Conroy J, Regan R, Fitzpatrick DJ, et al. (2012) HGDP and HapMap Analysis by Ancestry Mapper Reveals Local and Global Population Relationships. PLoS ONE 7(11): e49438.
II. QIAGEN’s Ingenuity Variant Analysis
Ingenuity Variant Analysis is a widely-used service used to analyze genomes by prioritizing variants through a series of cascading filters. CZ’s variants were ranked with the highest prioritized risk, based on their confidence, population frequencies, predictions of being deleterious, and comparison to known cancer drivers. At the end, these variants were associated with Epithelial and Head and Neck tumors (Figure 2).
Figure 2: Summary of Ingenuity’s Variant Interpretation of CZ’s genome. Filter cascades (left) show the removal of common and non-disease related variants, leaving a subset of 177 genes with 541 mutations that are related to cancer risk. The top-ranking cancers are epithelial and head and neck cancers.

III. Omicia and ClinVar for Clinical interpretation of risk and protective alleles (SNVs):
We next annotated variants using SnpEff software to identify variants in protein coding genes and assess impacts of variants on the protein product [1]. We further annotated these variants using the ClinVar database of pathogenic and non-pathogenic variants submitted through clinical channels on dbSNP and updated weekly; the version of the vcf format clinvar file used was from 03/02/2016 [2]. After filtering for quality and coverage (minimum quality score of 30 and minimum read depth of 10), we used these annotations to identify “high priority” variants, defining “high priority” as variants with moderate to high impact on protein sequence according to SnpEff and also present in the clinvar and OMIM (Online Mendelian Inheritance in Man) databases.
This identified 3,143 SNVs, of which 41 were marked by ClinVar as pathogenic and 5/41 SNVs as uncommon. These SNVs are in TERT, PRSS1 (2 SNVs), SAA1, and MEFV. Cross-referencing these SNVs with Omicia’s Opal Clinical software revealed that variants in MEFV lead to familial Mediterranean fever, but follow a recessive inheritance pattern, and since the variant identified was heterozygous, this SNV is thought to not have a phenotypic effect. The TERT variant (p.His412Tyr) is linked to shorter telomere lengths. PRSS1 variants are associated with hereditary pancreatitis, and the SAA1 variant (p.Ala70Val) may increase risk of reactive systemic amyloidosis (AA-amyloidosis) which is a rare complication of common chronic inflammatory diseases such as rheumatoid arthritis [3].
However, there were some mutations with good news. Specifically, mutations in C2 and CFB conferred a protection against age-related macular degeneration. Also, three mutations in IL23R revealed a lower risk of inflammatory bowel disease (IBD) and psoriasis (autoimmune). Moreover, protection against type II diabetes (MTTP), myocardial infarction and venus thrombosis (F13A1), and coronary heart disase (ABCA1) were all present. These indicate a genetic catalog of alleles with lower risk than the general population.
[1] P. Cingolani, A. Platts, L. L. Wang, M. Coon, T. Nguyen, L. Wang, S. J. Land, X. Lu, and D. M. Ruden, “A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3.,” Fly (Austin)., vol. 6, no. 2, pp. 80–92, Jan. .
[2] M. J. Landrum, J. M. Lee, G. R. Riley, W. Jang, W. S. Rubinstein, D. M. Church, and D. R. Maglott, “ClinVar: public archive of relationships among sequence variation and human phenotype.,” Nucleic Acids Res., vol. 42, no. Database issue, pp. D980–5, Jan. 2014.
[3] S. Baba, S. A. Masago, T. Takahashi, T. Kasama, H. Sugimura, S. Tsugane, Y. Tsutsui, and H. Shirasawa, “A novel allelic variant of serum amyloid A, SAA1 gamma: genomic evidence, evolution, frequency, and implication as a risk factor for reactive systemic AA-amyloidosis.,” Hum. Mol. Genet., vol. 4, no. 6, pp. 1083–7, Jun. 1995.
IV. Non-coding mutated genes
Beyond the protein-coding genes, there are many regulatory and non-coding genes across the genome that also can mediate disease or phenotypes. We scanned CZ’s genome for non-reference alleles and then intersected these variants with ENCODE regions and other regulatory regions such as VISTA enhancers. The transcription-factor binding sites with the most mutations were:
- STAT4 (Signal Transducer And Activator Of Transcription)
- AP1 (Activator Protein 1)
- EGR1 (nerve growth)
- AP2GAMMA1 (neural crest)
- GATA1 (hematopoietic TF)
There were also some VISTA enhancers (http://enhancer.lbl.gov) with an enrichment in mutations relative to the general population, and this included:
dorsal root ganglion, trigeminal V (ganglion, cranial)
2) element 2084
other
3) element 1760
heart
4) element 1886
heart
5) element 1385
hindbrain (rhombencephalon), midbrain (mesencephalon)
6) element 1391
midbrain (mesencephalon), forebrain
However, CZ’s body is already fully developed, so clearly these mutations did not prevent him from forming a complete midbrain, heart, or hindbrain. Which is good news, especially for CZ.
V. Transposable Element Insertions (TEIs)
We next used a computational tool called Jitterbug (https://github.com/elzbth/jitterbug) to scan for transposable element insertions (TEIs) that have inserted into CZ’s genome. We found 452 TEIs present in CZ’s genome, but all of these are also predicted from normal controls in the Pan-Cancer analysis of Whole Genomes (PCAWG) normal samples, n=64 controls), and thus they represent normal genetic variation.
Interestingly, 4 of these TEIs (Alus) are in introns, of the following genes:
MIR7641-2, RBM6, BRWD1, ENY2.
And 11 TEIs are in common with Genome in a Bottle GIAB) trio (son, father, mother):
HG002: 6, HG003: 5, HG004: 9
